The two most commonly used HPC applications in chemistry research on our campus are Gaussian ’09 (for ab initio and semi-empirical quantum mechanics), and NAMD (for classical molecular dynamics). To manage the queue of different computational jobs on the cluster, it makes use of the SLURM Workload Manager. In addition to the files containing your structure, parameters and/or topology, you will need an input script which essentially tells the HPC how much CPU time you are requesting, how many nodes and CPUs you want to use, as well as where the NAMD or Gaussian executables are.
Please contact dcashman@tntech.edu or the HPC cluster administrator, renfro@tntech.edu, if you have any questions or issues in running jobs on the HPC cluster.
Running a Gaussian ’09 Simulation
To initiate a job using Gaussian ’09, you will need Gaussian ’09 input file. This file usually ends with the suffix *.com and contains the method, basis set and internal coordinates of your system. If you used GaussView to generate this file, it will end with *.gjf, but the format is identical. More information on Gaussian input files is here (note that input for Gaussian ’09 and Gaussian ’16 use essentially the same commands).
You should upload all of the following files to the Tennessee Tech HPC cluster using the IP address provided to you by the HPC administrator (for security reasons, the IP address is not included here). Next, create a subdirectory in your home directory on the cluster for your simulation files to be contained in. Upload all simulation files to that directory using SCP. In that subdirectory, create a new file called g09_simulation.sh. This file should contain the following lines:
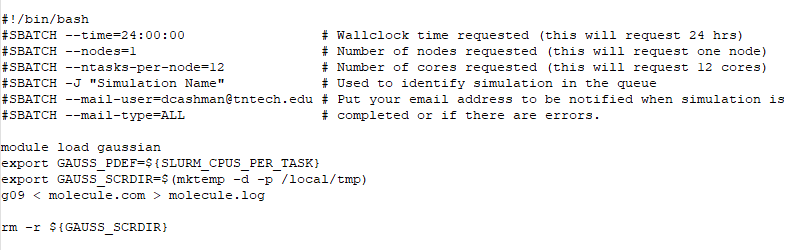
Save your simulation input file and ensure that the file is executable by typing chmod 755 g09_simulation.sh (or whatever your script is named). Please note that the cluster has 40 nodes with 28 CPU cores on each node. Gaussian ’09 does not scale well to multiple nodes or more than 12 or 16 CPUs. If you request more CPU cores than that, you may find that the software is not utilizing all cores (so you should not request more than 12 or 16 CPU cores).
To begin running your simulation, type sbatch g09_simulation.sh at the prompt. You may confirm the status of your job by typing squeue -u username. You may also view the HPC cluster queue from any workstation on campus at bright80.hpc.tntech.edu/slurm.
Note that some Gaussian ’09 calculations require more temporary scratch space on the hard disk (such as MP2 calculations). If you are having issues with jobs terminating because of lack of scratch space, you may wish to change the GAUSS_SCRDIR above a temporary directory in your home directory. To do so, type mkdir tmp in your home directory, and change the GAUSS_SCRDIR to ~username/tmp.
Running a NAMD Simulation
To initiate a simulation using NAMD, you will need the following structure/parameter files:
- PDB coordinate file of system of interest (if generated using MOE 2022, this file may end with *.coor)
- Protein Structure File (PSF) containing force field information
- Parameter file (PAR) containing all of the numerical information for evaluating forces and energies.
- NAMD Configuration File (conf) containing the parameters of the molecular dynamics simulation itself. If you’re using MOE 2022, this will be generated from the software itself. Please see the NAMD Tutorial for more information about what this file should contain.
You should upload all of the following files to the Tennessee Tech HPC cluster using the IP address provided to you by the HPC administrator (for security reasons, the IP address is not included here). Next, create a subdirectory in your home directory on the cluster for your simulation files to be contained in. Upload all simulation files to that directory using SCP. In that subdirectory, create a new file called namd_simulation.sh. This file should contain the following lines:
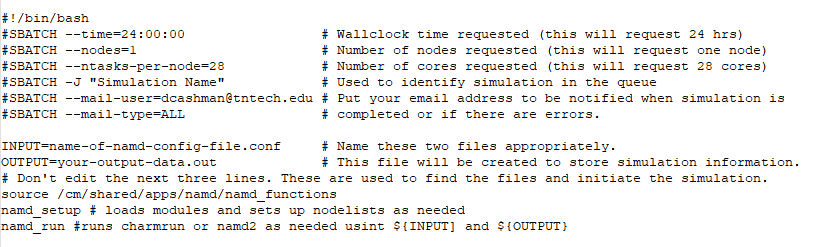
Save your simulation input file and ensure that the file is executable by typing chmod 755 namd_simulation.sh (or whatever your script is named). Please note that the cluster has 40 nodes with 28 CPU cores on each node. If you wish to run a simulation on multiple nodes, set (for example) nodes=2 to run on 56 CPU cores total (don’t change the ntasks-per-node variable).
To begin running your simulation, type sbatch namd_simulation.sh at the prompt. You may confirm the status of your job by typing squeue -u username. You may also view the HPC cluster queue from any workstation on campus at bright80.hpc.tntech.edu/slurm.
There is a second cluster containing a small number of AMD Ryzen CPUs. As this is not 100% in production yet, you should contact dcashman@tntech.edu if you are interesting in utilizing these resources.
Special Note on HPC Cluster Resources
Please note that this is a shared resources by many users throughout Tennessee Technological University. As such, you should be cognizant of that fact and not “hog” the system. While the maximum number of nodes you can request is 40, doing so will likely mean your job will be waiting in the queue a long time because you’re requesting 100% of the HPC cluster. If you do that, you will likely be contacted by the HPC Cluster administrator.
You should look at the current queue on the cluster before submitting any job and adjust your request for resources accordingly. As a general rule of thumb, Gaussian ’09 jobs should never be requesting more than 1 node or 16 CPU cores, and NAMD jobs should be limited to three to five nodes (84 to 140 CPU cores). You also may request more than 24 hours (several days of time is allowed). Be aware that the more resources that you request may result in a longer queueing time before your job runs because it may take longer for resources to be freed up by other jobs.