Introduction
This is a simple and fun, hands-on activity that demonstrates the basic concepts of computer-based molecular modeling. Using molecular mechanics, small molecules may be modeled by treating the atoms as balls and the bonds connected them as springs. We can then use a set of equations, which we refer to as a force field to place these atoms in the correct geometrical arrangement and calculate a relative energy that we can use to compare different conformations of these molecules.
The basic components of a typical molecular mechanics force field include:
Erelative = Ecovalent interactions + Enon-covalent interactions
The covalent and non-covalent interactions can further be broken down into the following components:
Ecovalent interactions = Ebond-stretching + Eangle-bending + Etorsion
Enon-covalent interactions = Eelectrostatic + Evan der Waal’s
The equations used for the covalent interactions are Hook’s law (spring harmonics). Equations used for the electrostatic interactions is Coulomb’s law, and the equations used for the van der Waal’s component is the 6-12 Lennard-Jones potential.
Materials
- Windows, Macintosh or Linux Notebook PC>
- Avogadro Molecular Modeling Software (available free of charge here)
- 3×5 Index Cards
- Sharpies or colored markers
Procedure
- Place one to three notebook computers on a table and have Avogadro already running as participants approach. It may also be advisable that the Desktop space is clear of files as well, in the event that Avogadro is closed or accidentally crashes during the dmeo and must be restarted.
- Decide ahead of time a small collection of potential models for participants to construct, and draw their names and chemical structures on the 3×5 index cards. The best molecules to build in this project are small organic molecules.Examples of good molecules to use for this demonstration include:
- Water (H2O)
- Acetylene (C2H2)
- Carbon Dioxide (CO2)
- Carbonic Acid (H2CO3)
- Ethane
- Ethanol (CH3CH2OH)
- Ethylene
- Glucose or Fructose (C6H12O6)
- Methane
- Propane
- Ribose
- 20 Amino Acids
- Palmitic Acid
- alpha-Linoleic Acid
- Allow participants a few minutes to play with the software and familiarize themselves with building small molecules. You may want to explain that the best way to build molecules is to start with their carbon chain, and build everything as if all atoms are carbons first. Then, modify specific atoms in functional groups to oxygen or nitrogen or whatever is necessary. Hydrogens do not need to be explicitly added (the software adds those automatically).
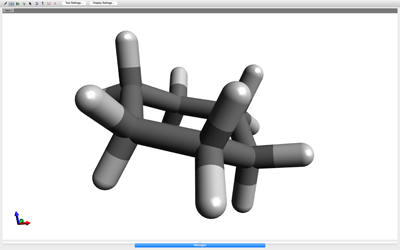
- Once a molecule is built on the screen, the molecule can be optimized to its correct geometry through a process called energy minimization. This process applies the force field and moves the atoms to their optimum positions based on the lowest energy conformation (you are minimizing the energy to its lowest point – hint: draw them an energy diagram as an example).Go to the EXTENSIONS > MOLECULAR MECHANICS menu option and select SETUP FORCE FIELD. Use the MMFF94 force field, change the number of steps to 2,000, change the algorithm to CONJUGATE GRADIENT, and the convergence criterion to 1 x 10-7, and press OK. Run the procedure by going to EXTENSION > OPTIMIZE GEOMETRY and the molecule should appear in its lowest energy conformation.If it does not appear in its lowest energy conformation, that means that the computer put the molecule into a LOCAL ENERGY MINIMA, as opposed to the GLOBAL ENERGY MINIMA. Molecular mechanics energy minimization can only lower energy, it can not increase energy – so you are stuck in a local energy minima. To get out of this, you need to physically move some atoms and re-minimize to recalculate the new energy.
- A simple experiment that can be done to illustrate the concept of minimization and local vs. global energy minima, is to have them build cyclohexane. This is easy to build as they just draw six carbon atoms and connect them all in a ring. Have them rotate this molecule around a bit to observe that the atoms are more or less arranged flat (not correct).Apply the energy minimization procedure, as specified above, to optimize the geometry. Most of the time, the computer will obtain the correct, “chair”, conformation of cyclohexane. Occasionally, some computers may minimize to the, “boat”, or, “twist-boat”, conformation – they are stuck in a local minima.See if the participant can convert between the boat and chair conformations of cyclohexane by moving atoms around and re-minimizing.
Safety
There are no safety issues with this demonstration.
Disposal of Waste Products
There are no waste disposal issues with this demonstration.